NiL2 Capillary
Capillary Laboratory Data on a NiL2 Coordination Complex
Aim of the exercise: The aim of this exercise is to illustrate a restrained Rietveld refinement of a relatively complicated molecular compound through the example of a Ni coordination complex. Laboratory X-ray diffraction data set have been collected in capillary mode using a Bruker D8 Advance diffractometer equipped with an incident beam monochromator and an mBraun linear PSD. The asymmetric unit actually contains two half molecules as shown in the figure below. The atomic numbering scheme in the figure will help you when you set up distance restraints. You’ll need the atom sequence number from the text list when you set up planar restraints.
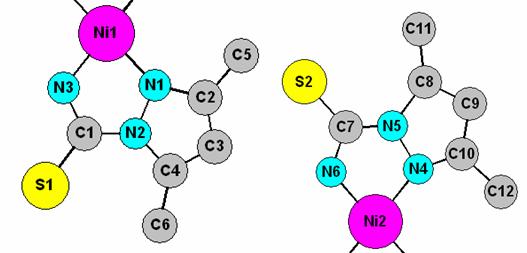
Starting files: d8_01914_sum.gsas; nil2.inst; nil2_start.cif
1. Ni1 Ni 0.0000 0.0000 0.0000 0.0200
2. Ni2 Ni 0.5000 0.5000 0.5000 0.0200
3. N1 N 0.0735 0.0765 0.2535 0.0200
4. N2 N 0.1800 0.0430 0.3670 0.0200
5. N3 N 0.1395 -0.0688 0.0650 0.0200
6. N4 N 0.5940 0.4330 0.2220 0.0200
7. N5 N 0.6860 0.4855 0.0865 0.0200
8. N6 N 0.6185 0.5854 0.3975 0.0200
9. C1 C 0.2080 -0.0395 0.2735 0.0200
10. C2 C 0.0810 0.1605 0.3500 0.0200
11. C3 C 0.1780 0.1755 0.4985 0.0200
12. C4 C 0.2432 0.1005 0.5190 0.0200
13. C5 C -0.0167 0.2225 0.2670 0.0200
14. C6 C 0.3565 0.0814 0.6665 0.0200
15. C7 C 0.7000 0.5670 0.1795 0.0200
16. C8 C 0.7795 0.4340 -0.0970 0.0200
17. C9 C 0.7310 0.3500 -0.0590 0.0200
18. C10 C 0.6270 0.3570 0.1360 0.0200
19. C11 C 0.8943 0.4517 -0.2740 0.0200
20. C12 C 0.5485 0.2813 0.2480 0.0200
21. S1 S 0.3220 -0.1085 0.4000 0.0200
22. S2 S 0.7780 0.6491 -0.0110 0.0200
Instructions
1. Start an EXP file. In tab “Phase”, read in cell parameters, space group and atoms from nil2_start.cif. You should read in 22 non-hydrogen atoms. Read in the diffraction pattern and instrument file.
2. Set the background function to type 1 and refine 15 terms. Rp ~17 %, RF2 ~ 25 % (16 pars).
3. Refine cell parameters, a single uiso on all atoms and the zero point. chi**2 ~10.4 (24 pars).
4. Refine profile parameters – use a damping factor of 5-9. chi**2 ~6.2.
5. Set up the necessary restraints for the refinement of atomic parameters by using the atomic numbering scheme shown above and the table below. You need to do this in the expedt routine within PC GSAS. (a) planar restraints: assume the whole ligand to be planar and introduce two planar restraints (one for each unique half-molecule) in: expedt>ls set up>soft constraints>planar. (b) bond length restraints: expedt>ls set up>soft constraints>bonds.
| Bond Length | Restraint | # found |
| Ni-N | 1.90(1) | 8 |
| C-C | 1.37(1) | 4 |
| C-N (in dimethylpyrazole ring) | 1.35(1) | 4 |
| N-N | 1.38(2) | 2 |
| C-CH3 | 1.49(1) | 4 |
| C-N (in thiocarbozamide C1 or C7) | 1.34(1) | 4 |
| C-S | 1.70(1) | 2 |
If you have input this correctly, you should have a total of 28 bond restraints. Increase the weight of constraints (planar and bond) to 20. This is high and will be decreased later, but it keeps the refinement from diverging the first time atomic parameters are allowed to vary.
6. Now refine atomic coordinates with a damping of 9. chi**2 ~4-5.
Outcome
This example should show you how careful you have to be when refining complex molecules from powder diffraction data. You should also realize that one would ideally want data of better quality than used in this example!
Additional Work (optional)
Try playing with weighting factors to see how they influence R-factors and the shape of the molecule. If you’re familiar with single crystal packages try introducing H atoms and let them ride on C atoms (they do have a significant influence on low angle reflections in this example).