Tutorial Organicriet
Organic Rietveld Refinement
Files needed: d8_00796.raw; rigidrod.cif
Learning Outcomes: This example shows a Rietveld refinement of an organic molecule using (bad) lab data. The use of restraints and rigid bodies is illustrated. Warning: some of the concepts here are relatively advanced! The molecule being used is:
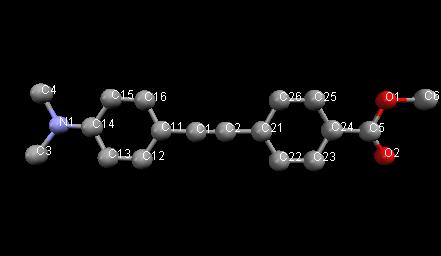
Topas advantages: lots of different ways of using restraints, rigid bodies, etc.
1. Save the datafile and cif file in your working directory.
2. Work through the menus in the Rietveld refinement section of Durham jedit menus.
3. Click on “Select Data File” and navigate to find the file d8_00796.raw.
4. In Instrument/Corrections select “Durham_d8” – the diffractometer used to collect the data.
5. Click on “Refine zero point” to flag that zero should be refined.
6. In “Structure – cif” click on the icon to “Read a CIF file” and select rigidrod.cif.
7. Set up to refine a single temperature factor on each atom (add “beq bvalue 1” at the end of each site line. Set to refine a, b and c cell parameters. Set to refine from 4 to 50 degrees (use start_X and finish_X icons in “Miscellaneous” section after putting the cursor immediately after the data filename).
8. You should be able to get Rwp=11.74%.
9. Before refining atomic coordinates it’s sensible to introduce restraints to prevent atoms refining to silly positions. Those listed below restrain benzene rings to be regular (via distances and angles) and planar (via the flatten restraint) and the NMe2 group on the molecule to be planar. Each macro contains the sites to be restrained, the ideal distance, the actual distance, a tolerance (difference before restraint applied) and a weight for the restraint.
‘restrain one benzene ring
Distance_Restrain(C11 C12, 1.400, 1.43463, 0, 1)
Distance_Restrain(C12 C13, 1.400, 1.41595, 0, 1)
Distance_Restrain(C13 C14, 1.400, 1.60138, 0, 1)
Distance_Restrain(C14 C15, 1.400, 1.45696, 0, 1)
Distance_Restrain(C15 C16, 1.400, 1.40217, 0, 1)
Distance_Restrain(C16 C11, 1.400, 1.49306, 0, 1)
Angle_Restrain(C11 C12 C13,120, 118.52024,0, .001)
Angle_Restrain(C12 C13 C14,120, 118.41604,0, .001)
Angle_Restrain(C13 C14 C15,120, 118.54301,0, .001)
Angle_Restrain(C14 C15 C16,120, 120.52919,0, .001)
Angle_Restrain(C15 C16 C11,120, 119.61364,0, .001)
Angle_Restrain(C16 C11 C12,120, 123.25791,0, .001)
Flatten(C11 C12 C13 C14 C15 C16,0, 0.0188811239,10)
‘restrain second benzene ring
Distance_Restrain(C21 C22, 1.400, 1.42104, 0, 1)
Distance_Restrain(C22 C23, 1.400, 1.37979, 0, 1)
Distance_Restrain(C23 C24, 1.400, 1.43837, 0, 1)
Distance_Restrain(C24 C25, 1.400, 1.39612, 0, 1)
Distance_Restrain(C25 C26, 1.400, 1.48327, 0, 1)
Distance_Restrain(C26 C21, 1.400, 1.38858, 0, 1)
Angle_Restrain(C21 C22 C23,120, 120.62386,0, .001)
Angle_Restrain(C22 C23 C24,120, 120.88302,0, .001)
Angle_Restrain(C23 C24 C25,120, 119.45380,0, .001)
Angle_Restrain(C24 C25 C26,120, 118.98380,0, .001)
Angle_Restrain(C25 C26 C21,120, 119.40741,0, .001)
Angle_Restrain(C26 C21 C22,120, 120.31336,0, .001)
Flatten(C21 C22 C23 C24 C25 C26,0, 0.0072359109,10)
‘restrain central linking group
Distance_Restrain(C21 C2 , 1.430, 1.60025, 0, 1)
Distance_Restrain(C11 C1 , 1.430, 1.40512, 0, 1)
Distance_Restrain(C2 C1 , 1.200, 1.27122, 0, 1)
‘restrain nme2 group
Distance_Restrain(C14 N1 , 1.380, 1.54348, 0, 1)
Distance_Restrain(N1 C3 , 1.450, 1.45761, 0, 1)
Distance_Restrain(N1 C4 , 1.450, 1.49031, 0, 1)
Flatten(N1 C3 C4 C14,0, 0.000726450936,10)
‘restrain CO2CH3 group
Distance_Restrain(C24 C5 , 1.500, 1.58680, 0, 1)
Distance_Restrain(C5 O1 , 1.350, 1.38465, 0, 1)
Distance_Restrain(C5 O2 , 1.200, 1.25119, 0, 1)
Distance_Restrain(O1 C6 , 1.440, 1.50432, 0, 1)
10. Flag all atomic coordinates to refine. You should get Rwp of 8.44%. Have a look at the ideal and restrained bond distances in the .inp file after refinement. Try using “view_structure” in your input file to look at the molecule. In case you mess up an example file is here.
11. We can try the same refinement using rigid bodies to describe the benzene rings. This can be done by introducing the lines below immediately below the last atomic coordinate line in the file:
rigid
Hexagon_sitting_on_side_in_xy_plane(C12, C13, C16, C15, C11, C14, 1.4)
Rotate_about_axies( @ 55.30755`, @ 117.44639`, @ 356.92214`)
Translate( @ 0.63680`, @ 0.48817`, @ 0.58321`)
rigid
Hexagon_sitting_on_side_in_xy_plane(C22, C23, C26, C25, C21, C24, 1.4)
Rotate_about_axies( @ 112.97294`, @ 244.56505`,@ -176.67930`)
Translate( @ 0.15133`, @ 0.50553`, @ 0.38959`)
The coordinates for atoms in benzene rings are now replaced by those of an ideal hexagon whose position and rotation is refined. Because atomic sites are no longer refined independently you must remove the “@” symbols from the coordinate lines of C11-C26. i.e. the file should look like:
site N1 x @ 0.85749` y @ 0.54252` z @ 0.65583` occ N 1 beq bvalue 3.0675`
site O1 x @ -0.29459` y @ 0.42989` z @ 0.30397` occ O 1 beq bvalue 3.0675`
site C1 x @ 0.43507` y @ 0.48261` z @ 0.50899` occ C 1 beq bvalue 3.0675`
site C2 x @ 0.37059` y @ 0.52090` z @ 0.47274` occ C 1 beq bvalue 3.0675`
site O2 x @ 0.03491` y @ 0.54861` z @ 0.28399` occ O 1 beq bvalue 3.0675`
site C3 x @ 1.02060` y @ 0.62899` z @ 0.67088` occ C 1 beq bvalue 3.0675`
site C4 x @ 0.70753` y @ 0.44862` z @ 0.68755` occ C 1 beq bvalue 3.0675`
site C5 x @ -0.07888` y @ 0.47581` z @ 0.31011` occ C 1 beq bvalue 3.0675`
site C6 x @ -0.39808` y @ 0.47302` z @ 0.26452` occ C 1 beq bvalue 3.0675`
site C11 x 0.53155 y 0.49281 z 0.54557 occ C 1 beq bvalue 3.0675`
site C12 x 0.73453 y 0.57636 z 0.55047 occ C 1 beq bvalue 3.0675`
site C13 x 0.83977 y 0.57172 z 0.58811 occ C 1 beq bvalue 3.0675`
site C14 x 0.74205 y 0.48354 z 0.62085 occ C 1 beq bvalue 3.0675`
site C15 x 0.53908 y 0.39999 z 0.61595 occ C 1 beq bvalue 3.0675`
site C16 x 0.43383 y 0.40463 z 0.57831 occ C 1 beq bvalue 3.0675`
site C21 x 0.24937 y 0.51019 z 0.42789 occ C 1 beq bvalue 3.0675`
site C22 x 0.36026 y 0.57890 z 0.39422 occ C 1 beq bvalue 3.0675`
site C23 x 0.26221 y 0.57423 z 0.35591 occ C 1 beq bvalue 3.0675`
site C24 x 0.05328 y 0.50086 z 0.35129 occ C 1 beq bvalue 3.0675`
site C25 x -0.05761 y 0.43216 z 0.38496 occ C 1 beq bvalue 3.0675`
site C26 x 0.04044 y 0.43682 z 0.42327 occ C 1 beq bvalue 3.0675`
rigid
Hexagon_sitting_on_side_in_xy_plane(C12, C13, C16, C15, C11, C14, 1.4)
Rotate_about_axies( @ 55.30755`, @ 117.44639`, @ 356.92214`)
Translate( @ 0.63680`, @ 0.48817`, @ 0.58321`)
rigid
Hexagon_sitting_on_side_in_xy_plane(C22, C23, C26, C25, C21, C24, 1.4)
Rotate_about_axies( @ 112.97294`, @ 244.56505`,@ -176.67930`)
Translate( @ 0.15133`, @ 0.50553`, @ 0.38959`)
When you refine you should be able to get Rwp=9.08%. Note that in the output file the restraints for the rings are now “perfect”. They’re redundant and could be deleted.
12. If you add a few extra background terms (say 6 more) you can get Rwp=6.6% and a reasonable looking fit. In case you mess up, look at the file linked here.